Phenylketonuria - العلامات الكلاسيكية للانتقال الوراثي والعلاج الغذائي
المحتويات
- 1كيف يتجلى فينيل كيتونوريا
- 2آلية تطور المرض
- 3فينيل كيتونوريا في الأطفال
- 4أعراض المرض
- 5الأسباب والمشغلات
- 6التشخيص
- 7علاج بيلة الفينيل كيتون الكلاسيكية
- 8ميزات التغذية لحديثي الولادة والعلاج الغذائي
- 9النظام الغذائي لأطفال ما قبل المدرسة وأطفال المدارس
- 10مجموعات المنتجات مع PKU
- 11كيفية التحكم في مستوى فينيل ألانين في الدم
- )12أشرطة الفيديو
الأمراض التي يرتبط حدوثها بعيوب في الجهاز الخلوي الوراثي - بيلة الفينيل كيتون - مدرجة في قائمة صغيرة من الأمراض الوراثية التي يمكن علاجها.كان رائد هذا المرض هو الطبيب النرويجي IA Felling ، وقد اكتشف لاحقًا أن جينًا واحدًا يسمى جين فينيل ألانين هيدروكسيلاز (الذراع الطويل للكروموسوم الثاني عشر الذي يحتوي على ما يصل إلى 4.5٪ من جميع مواد الحمض الخلوي الخلوي) كان مسؤولاً عن تطور ومجرى المرض.يؤدي الخلل الوراثي إلى تعطيل جزئي أو كامل لإنزيم الكبد فينيل ألانين-4-هيدروكسيلاز.
كيف يتم اكتشاف مرض بيلة فينيل الكيتون
يؤدي مرض بيلة فينيل الكيتون الموروثة (PKU) إلى تسمم مزمن في الجسم بمواد سامة تكونت نتيجة لضعف عملية التمثيل الغذائي للحمض الأمينيالهيدروكسيل من فينيل ألانين.التسمم الدائم يؤدي إلى تلف الجهاز العصبي المركزي (CNS) ، وهو مظهر من مظاهره انخفاض تدريجي في الذكاء (قلة فينيل بيروفيك).
يتجلى مرض فيلينج في التراكم المفرط في جسم الفينيل ألانين ومنتجات استقلابه الخاطئ.هناك عوامل أخرى في تطور بيلة الفينيل كيتون ، وتشمل ضعف نقل الأحماض الأمينية عبر الحاجز الدموي الدماغي ، انخفاض عدد الناقلات العصبية (السيروتونين ، الهستامين ، الدوبامين).في حالة عدم وجود علاج لهذا المرض في الوقت المناسب يؤدي إلى التخلف العقلي ويمكن أن يسبب وفاة الطفل.

آلية تطور المرض
سبب اضطرابات الجينات هو كتلة استقلابية تمنع تكوين فينيل ألانين-4-هيدروكسيلاز (إنزيم ،وهو المسؤول عن تحويل حمض أميني فينيل ألانين إلى تيروزين).يعتبر التيروزين الحمض الأميني البروتيني المنشأ جزءًا لا يتجزأ من البروتينات وصباغ الميلانين ، لذلك فهو عنصر ضروري لعمل جميع أجهزة الجسم ، ويؤدي افتقاره إلى اعتلال التخمر.
إن تثبيط تكوين المستقلب الناجم عن تعطيل التحور للإنزيم هو تنشيط مسارات التمثيل الغذائي المساعدة للفينيل ألانين.ينقسم حمض ألفا أمين العطري ، نتيجة لعمليات التمثيل الغذائي المعيبة ، إلى مشتقات سامة ، والتي لا تشكل في الظروف العادية:
- حمض فينيل بيروفيك (فينيل بيروفات) - حمض ألفا كيتو الدهني ، تكوينهيؤدي إلى النخاع من العمليات العصبية والخرف.
- حمض اللبنيك فينيل هو منتج يتكون أثناء استعادة حمض فينيل بيروفيك ؛
- فينيل إيثيل أمين - المركب الأولي لأجهزة الإرسال النشطة بيولوجيا للنبضات الكهروكيميائية ، يزيد من تركيز الدوبامين والأدرينالين والنورادرينالين ؛
- خلات الأرثوفينيل هو مادة سامة تسبب اضطرابات التمثيل الغذائي في الدماغ.
تشير الإحصاءات الطبية إلى أن الجين الذي تم تغييره بشكل مرضي موجود في 2٪ من السكان ، لكنه لا يظهر بأي شكل من الأشكال.ينتقل العيب الوراثي إلى الطفل من الوالدين فقط في وجود المرض لدى كلا الشريكين ، حيث يصبح الرضيع في 50٪ من الحالات حاملًا للجين المتحور ، ويبقى بصحة جيدة.احتمال أن يؤدي بيلة الفينيل كيتون في المواليد الجدد إلى 25٪.
حسب نوع الموروثة
مرض القطع هو خلل وراثي وراثي من نوع متنحي راثي.هذا النوع من الميراث يعني أن تطور علامات المرض الخلقي لن يحدث إلا عندما يرث الطفل الجين المعيب من كلا الوالدين حاملي الزيجوت غير المتجانسة للجين المعدل.
يحدث تطور مرض خلقي في 99 ٪ من الحالات بسبب طفرة في الجين المسؤول عن تشفير الإنزيم الذي يوفر تخليق فينيل ألانين - 4 - هيدروكسيلاز (فينيل كيتونوريا الكلاسيكي).ترتبط نسبة تصل إلى 1٪ من الأمراض الوراثية بالتغيرات الطفرية التي تحدث في الجينات الأخرى التي تسببهاقصور إنزيم ديهيدروبيريدين اختزال (PKU من النوع الثاني) أو رباعي هيدروبتيرين (PKU من النوع الثالث).
بيلة فينيل الكيتون في الأطفال
يظهر الشكل الكلاسيكي للأمراض الوراثية لدى الأطفال في معظم الحالات بشكل ملحوظ ظاهريًا ، بدءًا من 3-9 أشهر من العمر.الأطفال حديثي الولادة مع الجينات المعيبة ، تبدو صحية ، سمة مميزة هي العادة المحددة (المظهر) للطفل.تظهر الأعراض التي تم التعبير عنها خلال 6-12 شهرًا بعد الولادة.
يتميز النوع الثاني من PKU بحقيقة أن الأعراض السريرية الأولى تظهر بعد الولادة بسنة ونصف.لا تختفي علامات المرض بعد تشخيص التشوهات الوراثية وبداية العلاج الغذائي.غالبًا ما يؤدي هذا النوع من الأمراض الخلقية إلى نتيجة مميتة لمدة 2-3 سنوات من حياة الطفل.الأعراض الأكثر شيوعًا للنوع الثاني من PKU هي:
- اضطرابات عقلية شديدة ؛
- فرط المنعكسات ؛
- ضعف الحركة الحركية لجميع الأطراف ؛
- متلازمة تقلصات العضلات غير المنضبط.
تشبه العلامات السريرية للتغيرات الطفرية في جينات النوع الثالث مرض النوع الثاني.يتسم نقص رباعي هيدروبوبترين بثلاثة أعراض محددة:
- بدرجة عالية من التخلف العقلي ؛
- يتم تقليل حجم الجمجمة بوضوح مقارنة بأجزاء الجسم الأخرى ؛
- التشنج العضلي (الفقدان التام لحركة الأطراف ممكن).

مظاهر مرض القطع
في التجارب السريرية والملاحظات ، فقد اقترح أنتسبب المشتقات السامة لعملية التمثيل الغذائي للفينيل ألانين انخفاضًا في القدرة الذهنية ، وهي تقدمية في الطبيعة ويمكن أن تؤدي إلى الخرف (قلة القلة ، حماقة).من بين الأسباب المحتملة للاضطرابات التي لا رجعة فيها لنشاط الدماغ الأكثر منطقية أن يكون سببها انخفاض مستوى نقص التيروزين في الناقلات العصبية التي تنقل النبضات بين الخلايا العصبية.
لم يتم بعد تحديد العلاقة بين السبب والنتيجة الدقيقة بين المرض الوراثي واضطرابات الدماغ ، وكذلك آلية التطور بسبب بيلة الفينيل كيتون للحالات العقلية مثل الصدى ، الصدى ، النوبات الشرسة والتهيج.تشير نتائج التحليل إلى أن فينيل ألانين له تأثير سام مباشر على الدماغ ، مما قد يؤدي أيضًا إلى انخفاض في الذكاء.
الهيكل والسمات المظهرية
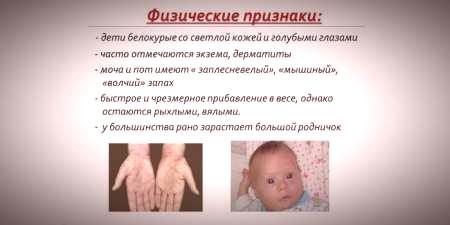
نظرًا لأن تشبع صبغة الجلد والشعر يعتمد على مستوى التيروزين في الميتوكوندريا في خلايا الكبد ، ويؤدي بيلة فينيل كيتون إلى توقف تحويل الفينيل ألانين ، فإن المرضى المصابين بهذا المرض لديهم خصوصياتعلامات متنحية).تؤدي زيادة لون العضلات إلى ظهور انحرافات في بنية الجسم - يصبح خلل التنسج.تشمل السمات الخارجية المميزة لداء بيلة الفينيل كيتون:
- نقص تصبغ الجلد - البشرة الفاتحة ، العيون الزرقاء الشاحبة ، الشعر المتغير.
- زرقة الأطراف ؛
- انخفاض حجم الرأس ؛
- وضع معين للجسم - عند محاولة الوقوف أو الجلوس ، يتبنى الطفل وضعية "خياط" (ثني الذراعين والساقين عند المفاصل).
الأعراضمرض
مع الكشف في الوقت المناسب ، يتم علاج مرض فينج بنجاح من خلال ضبط التغذية ويحدث نمو الطفل وفقا لفئته العمرية.تكمن صعوبة اكتشاف طفرة جينية في صعوبة اكتشاف العلامات المبكرة حتى من قبل طبيب أطفال متمرس.تزداد شدة أعراض المرض الخلقي مع تقدم الطفل في العمر ، لأن استهلاك البروتين الغذائي يسهم في تطور اضطرابات الجهاز العصبي المركزي.
علامات المواليد الجدد
خلال الأيام الأولى من حياة الطفل ، يصعب اكتشاف علامات التشوهات غير الطبيعية - يتصرف الطفل بشكل طبيعي ، ولا يوجد أي تأخير في النمو.تبدأ أعراض المرض في الظهور لأول مرة من 2-6 أشهر بعد الولادة.يجب أن يكون الوالدان متيقظين لسلوك الطفل الذي يتميز بانخفاض النشاط أو الخمول أو على العكس من القلق والإثارة المفرطة.[٩٠] (٩١) مع بداية الإرضاع من الثدي ، يبدأ المواليد الجدد مع الحليب في الدخول إلى جسم المولود الجديد مع الحليب ، وهو محفز لظهور العلامات الأولى ، والتي تشير بوضوح إلى أن المرض قد بدأ يتقدم.تشمل المظاهر السريرية المحددة للمرض ما يلي: [٩١] (٩٢) (٩٣) قيء مستمر (غالبًا ما يُعتبر ضيقًا خلقيًا لحارس المرمى) ؛
الأعراض عند الأطفال بعد 6 أشهر
إذا لم يكن مظهر المرض الوراثيحدث (أو لم يلاحظ) خلال الأشهر الستة الأولى بعد ولادة الطفل ، ثم بعد هذه الفترة أصبح من الممكن بالفعل تحديد الفارق الدقيق في النمو الحركي النفسي.أعراض الاضطرابات الوراثية الناجمة عن نقص الإنزيم في الأطفال الذين تزيد أعمارهم عن ستة أشهر هي:
- انخفاض النشاط (حتى إكمال اللامبالاة) ؛
- عدم ضبط النفس ، الجلوس ؛
- رائحة "ماوس" غريبة للجلد (رائحة العفن تنتج عن إفراز مشتقات فينيل ألانين السامة من خلال الغدد العرقية والبول) ؛
- فقدان القدرة على التعرف بصريًا على وجوه الوالدين ؛
- تقشير الجلد ؛
- التهاب الجلد ، الأكزيما ، تصلب الجلد.
تطور المرض في غياب العلاج في مرحلة الطفولة
إذا لم يتم اكتشاف تشوهات النمو في الطفولة ، ولم يتم تنفيذ العلاج المناسب ، ثميبدأ المرض في التقدم بنشاط وغالبا ما يؤدي إلى الإعاقة.يؤدي عدم العلاج في مرحلة مبكرة من المرض إلى ظهور الأعراض التالية عند عمر 1.5 عام:
- صغر الرأس (تقلص حجم المخ) ؛
- prognathia (تشريد الأسنان العلوية الأمامية) ؛
- التسنين المتأخر ؛
- نقص تنسج المينا (ترقق أو غياب كامل لمينا الأسنان) ؛
- التأخر في تطوير اللغة حتى الغياب التام للكلام ؛
- 3 ، 4 درجة من قلة القلة (التخلف العقلي ، التخلف العقلي) ؛
- عيوب القلب الخلقية (عيوب في بنية عضلة القلب ، وأجزاء من القلب ،سفن كبيرة) ؛
- اضطرابات في النظام اللاإرادي (تسمم الدم ، التعرق ، انخفاض ضغط الدم الشرياني) ؛
- الإمساك.
الأسباب والعوامل المحفزة
لإظهار طفرة جسمية متنحية ، يجب أن يرث جين معيب من كلا الوالدين.تحدث الأمراض الوراثية من هذا النوع بنفس التردد لدى الأولاد والبنات حديثي الولادة.يحدث التسبب في PKU بسبب ضعف التمثيل الغذائي للفينيل ألانين ، والذي يمكن أن يحدث في 3 أشكال.يخضع العلاج الغذائي فقط لنوع بيلة الفينيل كيتون الكلاسيكية
يمكن علاج أشكال غير نمطية من المرض عن طريق ضبط التغذية.هذه الانحرافات ناتجة عن نقص رباعي هيدروبتيرين ، اختزال ديهيدروبتيرين (نادراً - سينسيز بيروفيل تيتراهيدروبتيرين ، غوانوزين-5-فوسفات وغيرها).يتم تسجيل معظم حالات الحالات المميتة بين المرضى الذين يعانون من اختلافات نادرة من PKU ، مع المظاهر السريرية لجميع أشكال المرض مماثلة.يزداد خطر إنجاب طفل مع جين متحور من فينيل ألانين هيدروكسيلاز إذا كان الوالدان أقرباء (في حالات زواج وثيق الصلة).
التشخيص
في حالة الاضطرابات الوراثية المشتبه بها ، يتم التشخيص على أساس مجموعة من البيانات التي تم الحصول عليها من دراسة التاريخ الطبي - بيانات الأنساب ، ونتائج الدراسات الوراثية السريرية والطبية.للكشف في الوقت المناسب عن الأمراض الخلقية (PKU ، والتليف الكيسي ، galactosemia ، الخ) تم تطوير برنامج الكتلة الإجباريةالفحص المختبري لجميع المواليد الجدد (فحص حديثي الولادة).
إذا كان الوالدان الحاملان على دراية بحامل الجين المتحور ، فإن الطب الحديث يوفر طرقًا لاكتشاف خلل في مرحلة الحمل (تشخيص الجنين قبل الولادة بالطريقة الغازية).لتقسيم بيلة الفينيل كيتون إلى أنواع حسب الشدة ، يتم استخدام تصنيف مشروط يعتمد على مستوى فينيل ألانين في سائل بلازما الدم الخالي من الفيبرينوجين:
- بيلة فينيلكتونية ثقيلة - 1200 ميكرولول /لتر.
- متوسط - 60-1200 ميكرول /لتر.
- الضوء (لا يلزم العلاج) - 480 ميكرول /لتر.
اختبار الفحص
يحدث اكتشاف التشوهات الوراثية في عدة مراحل.في المرحلة الأولى في مستشفى الولادة ، يتم أخذ عينات من الأطفال الرضع لمدة تتراوح من 3-5 أيام إلى الدم المحيطي (من خمسة) للبحث.يتم تطبيق المادة على شكل ورقة وإرسالها إلى مختبر الكيمياء الحيوية حيث يتم تحليل الكيمياء الحيوية.في المرحلة الثانية من اختبار الفحص ، يتم تحديد تركيز تركيز فينيل ألانين الطبيعي.

إذا لم يتم اكتشاف أي تغييرات مرضية ، يتم الانتهاء من التشخيص وتسجيل بطاقة الطفل.في حالة الانحراف عن المعيار ، يتم إرسال نتائج التشخيص إلى طبيب الأطفال لضمان فحص أكثر دقة لعينة دم الوليد.تعتمد صحة الطفل على التنفيذ الدقيق وفي الوقت المناسب لجميع التدابير لاكتشاف الانحرافات.إذا تم تأكيد التشخيص بعد الفحص المتكرر ، فإن والدي الطفل سوفإرسالها إلى عيادة وراثة الأطفال لتلقي العلاج.
تحليلات ودراسات لتأكيد التشخيص
يتم إجراء إعادة التشخيص عند الكشف خلال اختبار الفحص الأساسي للتشوهات عن طريق إعادة تقديم الاختبارات.بالإضافة إلى تحديد محتوى فينيل ألانين في الدم لطرق تشخيص PKU لدى الأطفال والبالغين ، تشمل ما يلي:
- اختبار القطع - تحديد حمض فينيل بيروفيك في البول عن طريق إضافة كلوريد الحديد إلى المادة الحيوية (تلطيخ في اللون الأزرق والأخضر) ؛
- اختبار جوثري - تقييم درجة استجابة الكائنات الحية الدقيقة لعملية التمثيل الغذائي أو الإنزيمات الموجودة في دم المريض ؛
- تحليل كروماتوجرافي - دراسة الخواص الكيميائية للمواد الموزعة بين مرحلتين ؛
- قياس الفلور - تشعيع المادة الحيوية بواسطة الإشعاع أحادي اللون لتحديد تركيز المواد الواردة فيه ؛
- تخطيط كهربية الدماغ - تشخيص النشاط الكهربائي للدماغ ؛
- التصوير بالرنين المغناطيسي هو إثارة النواة الذرية للخلايا بواسطة الموجات الكهرومغناطيسية وقياس استجابتها.
علاج بيلة الفينيل كيتون الكلاسيكية
في قلب علاج بيلة الفينيل كيتون ، هناك قيود على استهلاك المنتجات التي تعتبر مصدرًا للبروتينات النباتية والحيوانية.الطريقة الوحيدة للعلاج الناجح هي العلاج بالنظام الغذائي ، حيث يتم تقييم مدى كفاية محتواه من فينيل ألانين في المصل.أقصى مستوى للأحماض الأمينية في المرضى من مختلف الفئات العمريةهو:
- عند الرضع والأطفال حتى عمر 3 سنوات - ما يصل إلى 242 ميكرولتر /لتر ؛
- في رياض الأطفال ما يصل إلى 360 ميكرول /لتر ؛
- في المرضى الذين تتراوح أعمارهم بين 7 و 14 عامًا - ما يصل إلى 480 ميكرول /لتر ؛
- لدى المراهقين - ما يصل إلى 600 ميكرول /لتر.
تعتمد فعالية النظام الغذائي على مرحلة المرض التي يتم تصحيح النظام الغذائي لها.في التشخيص المبكر للعلاج المرضي الخلقي يوصف العلاج من الأسبوع الثامن من الحياة (بعد هذه الفترة تبدأ تغييرات لا رجعة فيها بالفعل).يؤدي غياب التدابير في الوقت المناسب إلى مضاعفات وانخفاض مستوى الذكاء بمقدار 4 نقاط لمدة شهر واحد من الولادة وحتى بداية العلاج.

بالنظر إلى أن النظام الغذائي العلاجي لفينيل كيتونوريا يوفر الاستبعاد التام من نظام البروتين الحيواني ، هناك حاجة إلى استخدام مصادر أخرى من الأحماض الأمينية الأساسية ، وكذلك فيتامينات المجموعة ب ، الكالسيوم -والمركبات المعدنية المحتوية على الفوسفور.تشمل المنتجات التي يجب استخدامها كمكملات خالية من البروتين ما يلي:
- هيدروليات البروتين (Amigen ، Aminazole ، Fibrinosol) ؛
- لا تحتوي على مخاليط فينيل ألانين مشبعة بالأحماض الأمينية الأساسية - خالية من التترافين ، خالية من الفينيل.
بالإضافة إلى التدابير العلاجية للقضاء على سبب ضعف أداء الجسم ، يجب إجراء علاج الأعراض بهدف القضاء على عيوب النطق وتنسيق الحركات بشكل طبيعي.يشمل العلاج المركب إجراءات العلاج الطبيعي والتدليك ومساعدة أخصائي علاج النطق وأخصائي نفسي وأداء تمارين رياضية.في بعض الحالات ، بالتزامن مع العلاج الغذائييظهر استخدام مضادات الاختلاج والعقاقير منشط الذهن والأوعية الدموية.
لا يمكن علاج خصوصيات علاج الأشكال غير النمطية
النوع الثاني والثالث من الفينيلكتونينية مع اتباع نظام غذائي منخفض البروتين - يظل مستوى الفينيل ألانين في الدم دون تغيير مع الحد من تدفق البروتين إلى الجسم أو الأعراض السريرية حتى مع انخفاض مستوى الأحماض الأمينية.يتم إجراء علاج فعال لهذه الأشكال من المرض باستخدام:
- رباعي هيدرو بيو ترين - عامل الإنزيم المصاب ؛
- نظائرها الاصطناعية من رباعي هيدروبتيرين - هذه المواد تخترق بشكل أفضل حاجز الدم في الدماغ ؛
- عقاقير علاج الإحلال - لا تقضي على سبب بيلة الفينيل كيتون ، ولكن تدعم الأداء الطبيعي للجسم (ليفودوبا مع كاربيدوفا ، 5 أكسي تريبتوفان ، 5 فورميل تيتراهيدروفولات) ؛
- حماية الكبد - دعم وظائف الكبد ؛
- مضادات الاختلاج ؛
- إدخال جين فينيل ألانين هيدروكسيلاز في الكبد هو طريقة تجريبية.
التغذية والعلاج بالمواليد حديثي الولادة
يُسمح بالرضاعة الطبيعية في السنة الأولى من عمر الطفل المصاب بمرض PKU ، ولكن يجب تقييده.يصل إلى 6 أشهر ، الكمية المقبولة من فينيل ألانين هي 60-90 ملغ لكل 1 كجم من وزن الطفل (100 غرام من الحليب يحتوي على 5.6 ملغ من فينيل ألانين).من 3 أشهر ، يجب توسيع نظام الطفل الغذائي تدريجياً ، مع إدخال عصائر الفاكهة وهريسها.
يُسمح للأطفال من عمر 6 أشهر بوضع نظام غذائي من المهروسات النباتية ، عصيدة (ساغو) ، خالية من البروتينكيسيليف.بعد 7 أشهر ، يمكن إعطاء معكرونة منخفضة البروتين للأطفال ، ابتداء من 8 أشهر - خبز لا يحتوي على بروتين.لم يتم تحديد العمر الذي يجب فيه تقييد البروتين في جسم الطفل المريض.لا يزال الأطباء يناقشون جدوى علاج النظام الغذائي مدى الحياة ، لكنهم يتفقون على ضرورة اتباع نظام غذائي لا يقل عن 18.
لا يُعد تشخيص بيلة الفينيل كيتون في المرأة سببًا لرفض ولادة طفل.الأمهات الحوامل المصابات بـ PKU لمنع إصابة الجنين أثناء الحمل ولمنع المضاعفات المحتملة ، من الضروري مراقبة نظام غذائي مع اتباع نظام غذائي مقيد بالفينيل ألانين (في وقت الولادة) (حتى 242 ميكروليتر /لتر).
خلطات خالية من اللاكتوز للأطفال
يعتمد نظام غذائي لفينيل الكيتون على انخفاض كبير في كمية البروتين الطبيعي في النظام الغذائي اليومي ، لكن جسم الرضيع لا يمكن أن يتطور بشكل طبيعي في غياب العناصر النزرة الضرورية.لتلبية حاجة الطفل للبروتين ، يتم استخدام مخاليط الأحماض الأمينية الخالية من اللاكتوز ، والتي بموجب القانون الروسي ، يجب توفيرها مجانًا.
يتغير تسامح الرضيع مع فينيل ألانين بسرعة خلال السنة الأولى من العمر ، لذلك يجب مراقبة تركيزه في دم الطفل وتعديل النظام الغذائي.تم تصميم المخاليط لفئات عمرية معينة:
- يتم وصف الأطفال دون سن عام Afenilak 15 و Analog-SP و PKU-1 و PKU-mix و PKU Anamix ؛
- طفل أكبر من عمر واحدفي السنة ، قم بتعيين المخصب بالفيتامينات ومخاليط المعادن التي تحتوي على نسبة عالية من البروتين - PKU Prima ، P-AM Universal ، PKU-1 ، PKU-2 ، Maximeid XP ، Maximum XP.

الأطعمة المكملة للبروتين
أحد المكونات الرئيسية لنظام غذائي لفينيل كيتونوريا هو المنتجات القائمة على النشا منخفض البروتين.تحتوي هذه المكملات على الكازين هيدرليزيت ، التربتوفان ، التيروزين ، الميثيونين ، النيتروجين وتوفر متطلبات الطفل اليومية للبروتين اللازم للتطور والنمو الطبيعي.الأطعمة المتخصصة التي تملأ نقص المعادن الأساسية والأحماض الأمينية عند نقصها في النظام الغذائي هي:
- بيرلوفن ؛
- زيمورغان ؛
- مينافن ؛
- أبونتي.
النظام الغذائي لمرحلة ما قبل المدرسة وتلاميذ المدارس
نظرًا لأن الجسم يتكيف مع الفينيل ألانين ، يمكن للأطفال من سن 5 سنوات تقليل قيودهم الغذائية تدريجياً.يتم توسيع النظام الغذائي عن طريق إدخال الحبوب ومنتجات الألبان ومنتجات اللحوم.الطلاب كبار لديهم بالفعل التسامح عالية للفينيل ألانين ، لذلك في هذا العمر يمكنك الاستمرار في توسيع النظام الغذائي ، مع مراقبة الاستجابة لأي تغييرات في التغذية.تستخدم الطرق التالية للسيطرة على حالة الطفل:
- تقييم المعلمات العصبية ، الحالة النفسية ؛
- التحكم في أداء مخطط كهربية الدماغ ؛
- تحديد مستوى فينيل ألانين.
مجموعات من الأطعمة مع PKU
نظام غذائي لمرضى PKU ، جنبا إلى جنب مع انخفاض البروتينتشمل المنتجات النشوية والمخاليط الطبية أيضًا منتجات ذات أصل طبيعي.عند إعداد القائمة ، يجب أن تحسب كمية البروتين المستهلكة بشكل واضح ويجب ألا تتجاوز الجرعة التي أوصى بها الطبيب.للتخلص من الآثار السامة على الجسم ، تم تطوير 3 قوائم من المنتجات تحتوي على مواد محظورة (حمراء) ، غير موصى بها (برتقالية) ومسموح بها (خضراء).
القائمة الحمراء
تطور بيلة فينيل كيتونينية على خلفية عدم وجود إنزيم يتحول إلى التيروزين فينيل ألانين ، لذلك فإن محتوى البروتين المرتفع هو الأساس لإدراج المنتجات في القائمة المحظورة (الحمراء).يجب أن تستبعد العناصر الموجودة في هذه القائمة تمامًا حمية مريض PKU:
- من اللحوم ؛
- الأعضاء الداخلية للحيوانات ، والمنتجات الثانوية ؛
- النقانق ، النقانق.
- المأكولات البحرية (بما في ذلك الأسماك) ؛
- بيض من جميع الطيور ؛
- منتجات الألبان ؛
- جوز ؛
- من البقول والحبوب ؛
- منتجات الصويا ؛
- أطباق تحتوي على الجيلاتين ؛
- صناعة الحلويات ؛
- الأسبارتام.
القائمة البرتقالية
المنتجات المراد تناولها لطفل تم تشخيصه بـ PKU مدرجة في القائمة البرتقالية.إدراج العناصر الموجودة في هذه القائمة في النظام الغذائي أمر مقبول ، ولكن بعدد محدود للغاية.على الرغم من أن هذه المنتجات لا تحتوي على الكثير من البروتين ، إلا أنها يمكن أن تزيد من مستوى فينيل ألانين ، لذلك لا ينصح باستخدامها:
- خضروات معلبة ؛
- أطباق البطاطا والأرز ؛
- ملفوف ؛
- حليب ؛
- شربات.
القائمة الخضراء
يُسمح باستخدام المنتجات الخالية من البروتين في المرضى الذين يعانون من تشخيص بيلة الفينيل كيتون دون قيود.قبل شراء العناصر الموجودة في القائمة الخضراء ، من الضروري فحص التركيبة الموضحة على العبوة والتأكد من عدم وجود صبغة للأسبارتام المحتوية على فينيل ألانين:
- ثمار ؛
- خضروات (باستثناء البطاطا والملفوف) ؛
- التوت ؛
- أخضر ؛
- حبوب نشوية (ساغو) ؛
- عسل ، سكر ، مربى ؛
- منتجات دقيق مصنوعة من دقيق الذرة أو الأرز ؛
- زيوت ودهون (زبدة ، عباد الشمس ، زيتون).
كيفية التحكم في مستوى الفينيل ألانين في الدم
بيلة فينيل كيتون هو مرض غير قابل للشفاء يمكن نقله إلى مرحلة الركود من خلال استخدام النظام الغذائي والتدابير العلاجية.عند تغيير الظروف المعيشية ، يمكن أن يتفاقم اضطراب النظام الغذائي مرة أخرى ، وبالتالي يحتاج المرضى إلى مراقبة مدى الحياة.تتمثل عملية التحكم في تحديد مستوى فينيل ألانين في الدم بشكل دوري.يعتمد تكرار الفحص على عمر المريض:
- حتى 3 أشهر - يجب إجراء فحص الدم أسبوعيًا للحصول على نتائج متسقة ؛
- من 3 أشهر إلى سنة - 1-2 مرات في الشهر ؛
- 1 إلى 3 سنوات - مرة واحدة كل شهرين ؛
- أكبر من 3 سنوات - ربع سنوي.

يتم إعطاء الدم للتحليل بعد 3-4 ساعات من الابتلاع.بالإضافة إلى الفحص ، يتم التحكم في تطوير PKU من خلال تحديد الحالة التغذوية والجسدية والعاطفية للمريض ، ومستوى القدرات الفكرية.وتنمية اللغة.قد تتطلب الملاحظات تشخيصات إضافية بمشاركة متخصصين مناسبين.
الفيديو
المعلومات المقدمة في هذه المقالة هي للإرشاد فقط.المقالة لا تدعو إلى العلاج الذاتي.يمكن للطبيب المؤهل فقط تشخيص وتوصية العلاج على أساس الخصائص الفردية للمريض معين.